R Package for preprocessing, normalizing, and analyzing proteomics data
Introduction
High-throughput omics data are often affected by systematic biases introduced throughout all the steps of a clinical study, from sample collection to quantification. Failure to account for these biases can lead to erroneous results and misleading conclusions in downstream analysis. Normalization methods aim to adjust for these biases to make the actual biological signal more prominent. However, selecting an appropriate normalization method is challenging due to the wide range of available approaches. Therefore, a comparative evaluation of unnormalized and normalized data is essential in identifying an appropriate normalization strategy for a specific data set. This R package provides different functions for preprocessing, normalizing, and evaluating different normalization approaches. Furthermore, normalization methods can be evaluated on downstream steps, such as differential expression analysis and statistical enrichment analysis. Spike-in data sets with known ground truth and real-world data sets of biological experiments acquired by either tandem mass tag (TMT) or label-free quantification (LFQ) can be analyzed.
Installation
To install the package, run:
# Install PRONE.R from github and build vignettes
if (!requireNamespace("devtools", quietly = TRUE)) install.packages("devtools")
devtools::install_github("lisiarend/PRONE", build_vignettes = TRUE, dependencies = TRUE)
# Load and attach PRONE
library("PRONE")
# TODO: bioconductor installationDue to current issues with DEqMS on Bioconductor and the ongoing submission of PRONE to Bioconductor, the main branch of PRONE no longer includes DEqMS functions. If you need to perform DEqMS, please use the with_DEqMS branch.
# Install PRONE.R from github and build vignettes
if (!requireNamespace("devtools", quietly = TRUE)) install.packages("devtools")
devtools::install_github("lisiarend/PRONE", build_vignettes = TRUE, dependencies = TRUE, ref = "with_DEqMS")Workflow
A six-step workflow was developed in R version 4.2.2 to evaluate the effectiveness of the previously defined normalization methods on proteomics data. The workflow incorporates a set of novel functions and also integrates various methods adopted by state-of-the-art tools.
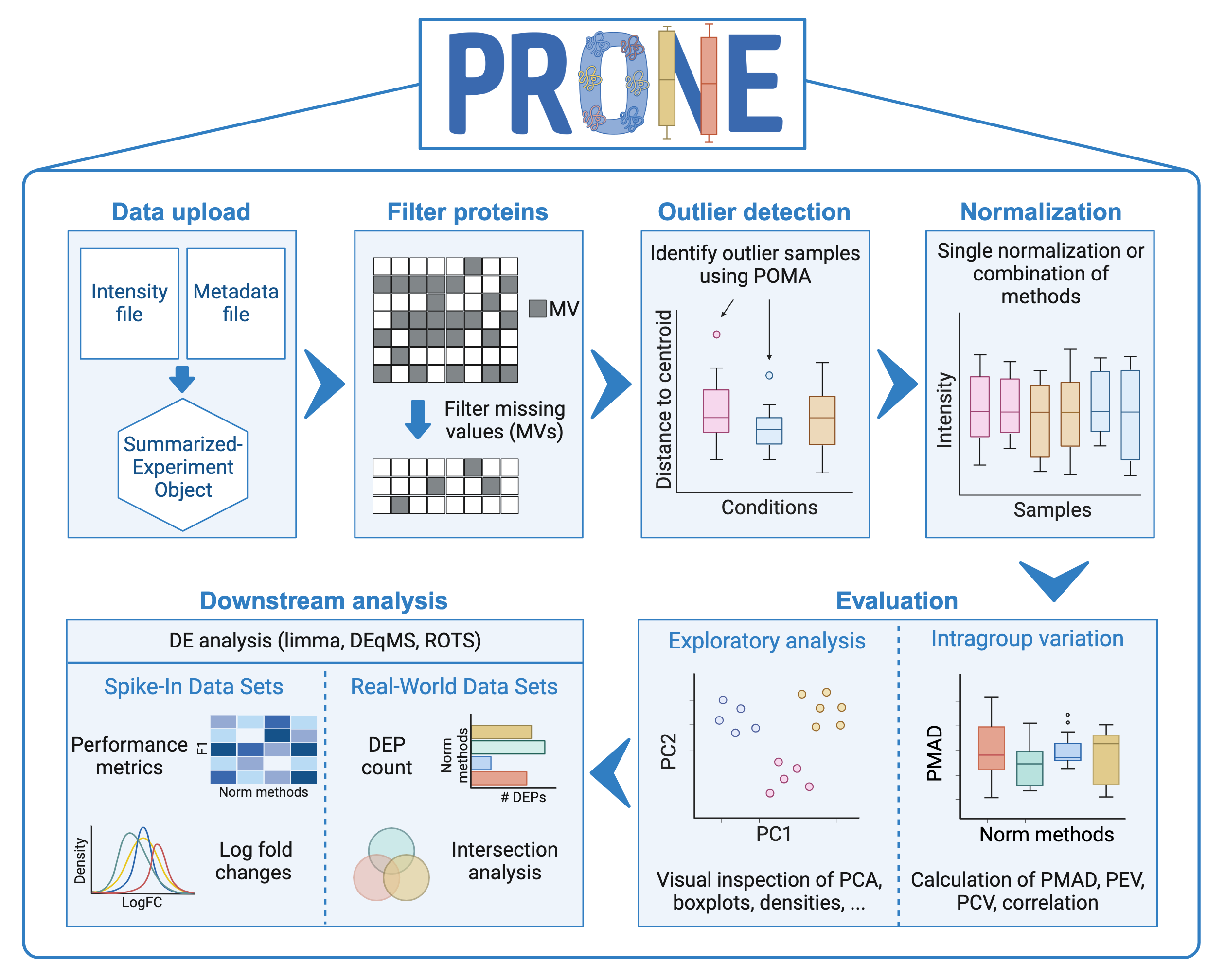
Following the upload of the proteomics data into a SummarizedExperiment object, proteins with too many missing values can be removed, outlier samples identified, and normalization carried out. Furthermore, an exploratory analysis of the performance of normalization methods can be conducted. Finally, differential expression analysis can be executed to further evaluate the effectiveness of normalization methods. For data sets with known ground truth, such as spike-in and simulated data sets, performance metrics, such as true positives (TPs), false positives (FPs), and area under the curve (AUC) values, can be computed. The evaluation of DE results of real-world experiments is based on visual quality inspection, for instance, using volcano plots, and an intersection analysis of the DE proteins of different normalization methods is available.
Usage
To get familiar with the functionalities of the R package, check out the article Getting started with PRONE.